

蛋白质功能预测的新SOTA技术:上海理工大学、牛津大学等机构采用基于统计的AI方法,研究成果发表于《自然》子刊。
编辑日期:2024年08月22日

编辑 | KX
蛋白质通过与其他分子相互作用,推动几乎所有基本的生物学过程。因此,理解蛋白质的功能对于揭示健康状态、疾病发展、生物进化以及生物体在分子层面的工作原理极其重要。
然而,目前仍有超过2亿种蛋白质未被充分表征,而现有的计算方法大多依赖于蛋白质结构信息来进行功能预测。
最近,由牛津大学、苏黎世联邦理工学院、上海理工大学及北京师范大学的研究人员组成的团队开发了一种基于统计的图网络方法——PhiGnet,该方法能够有效推进蛋白质功能注解和功能位点的识别。
PhiGnet不仅在性能上超越了其他现有方法,并且即便在缺乏结构信息的情况下也能显著缩小序列与功能之间的差距。研究表明,将深度学习应用到进化数据中可以精确地识别出残基级别的功能位点,这对于解析和探索蛋白质在生物医学领域的既有特性及新兴功能提供了有力的支持。
这项研究成果以“使用统计信息增强的图网络准确预测蛋白质功能”为题,在8月4日发表于《自然通讯》杂志上。

深入了解蛋白质的功能对于揭示众多关键生物学活动背后的复杂机制至关重要,并对医学、生物技术和药物研发等领域产生了深远影响。
截至2023年6月,UniProt数据库中已经收录了超过3.56亿种蛋白质序列,其中大约80%尚未获得明确的功能注解。
深度学习技术在预测蛋白质三维结构方面的精确度取得了显著突破,其表现已经超越了传统的从头计算法和同源性建模方法。然而,在准确地为蛋白质分配功能注解上仍面临挑战,尤其与实验测定结果相比。为了克服这些难题,研究者们认为可以从共同进化的残基中提取信息,用于残基层面的功能注解。
来自牛津大学的研究团队提出了一种基于统计的图网络方法,仅通过蛋白质序列就能预测其功能。这种方法自然地包含了进化的特征,并能定量评估执行特定功能的残基的重要性。
该方法利用从进化数据中获取的知识来驱动两个堆叠的图卷积网络。结合这些知识和定制的网络架构,能够准确地为蛋白质分配功能注解,并且重要的是,还能量化每个残基对于特定功能的重要程度。
PhiGnet 方法采用基于统计的图网络来注解蛋白质的功能,并依据其序列识别跨物种的功能位点。
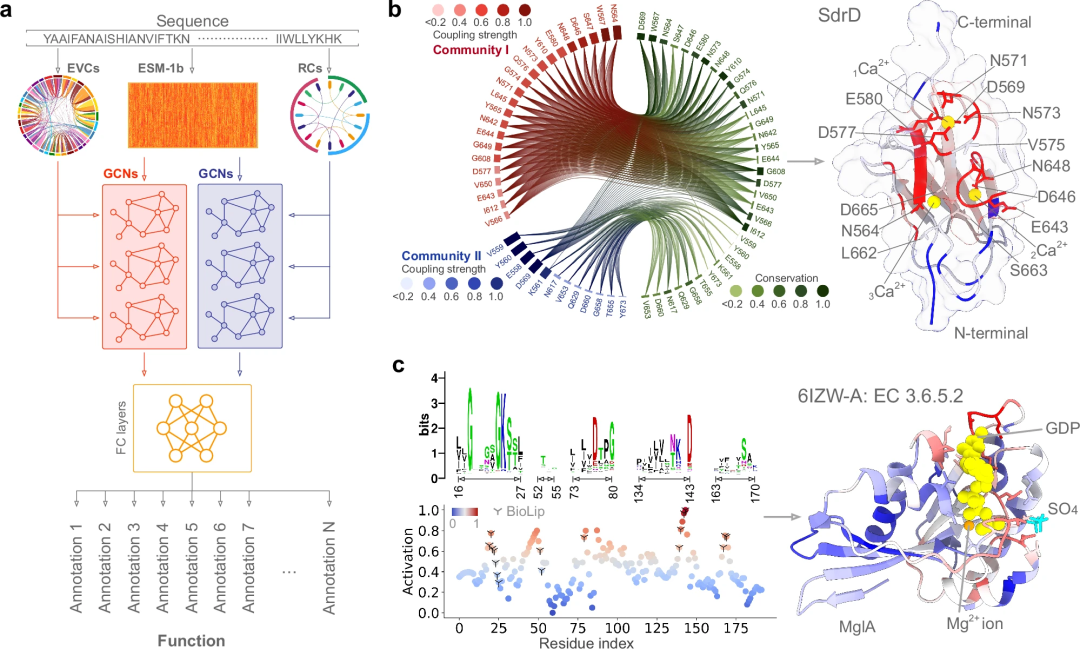
为了整合进化耦合(EVC,即两个共变异位点间的残基对关系)和残基群落(RC,残基间的层级相互作用)中的知识,研究者们设计了一个双通道架构,采用了堆叠图卷积网络 (GCN)。这一方法专门用于为蛋白质分配功能注解,包括酶委员会 (EC) 编号和基因本体 (GO) 术语(如生物过程、BP、细胞成分、CC 和分子功能、MF)。
当输入蛋白质序列时,研究者使用预训练的ESM-1b模型推导出其嵌入向量。随后,将这些嵌入向量作为图的节点,同时将EVC和RC(图的边)输入到双堆叠GCN的六个图卷积层中。这些层与两个全连接 (FC) 层协同工作,精心处理来自两个GCN的信息,最终生成一个概率张量,以评估为蛋白质分配功能注解的可能性。
此外,我们运用梯度加权类激活图 (Grad-CAM) 方法得到的激活分数来评估每个残基在特定功能中的重要性。这些分数使 PhiGnet 能够精确地定位到单个残基级别的功能位点。例如,在计算含有丝氨酸-天冬氨酸重复序列的蛋白质 D (SdrD) 的远程通讯(RC)时,我们发现功能关键的残基通过自然进化得以保存,PhiGnet 能够捕捉这类信息,从而改进在无结构数据情况下预测蛋白质功能的能力。
为了验证 PhiGnet 预测的准确性是否与实验确定的功能注释相当,研究者们利用激活分数定量分析了每种氨基酸对蛋白质功能的贡献。通过对九种蛋白质中残基重要性的评估,检验了 PhiGnet 的预测性能及其对蛋白质功能贡献的评估能力。
通过对这九种蛋白质中每个残基的激活分数进行计算,并与实验或半手动注释确定的关键残基进行对比,发现 PhiGnet 在预测残基级别的重要位点方面表现出良好的准确性(平均 ≥ 75%),并且与实际的配体/离子/DNA 结合位点高度一致。PhiGnet 成功地识别了具有高激活分数的蛋白质功能关键残基。
为了进一步评估 PhiGnet 的预测性能,我们将该方法应用于两个基准测试集中的蛋白质功能注释推断(EC 编号和 GO 术语)。与最先进的方法(包括基于比对的方法和基于深度学习的方法)进行比较。我们使用了两个核心指标来进行评估:以蛋白质为中心的 Fmax 分数和精确召回曲线下面积 (AUPR)。
PhiGnet 展示了其在两个测试集上为蛋白质分配功能注释的预测能力。它在GO术语和EC编号上分别达到了平均AUPR 0.70和0.89,以及Fmax分数0.80和0.88。总体来说,PhiGnet在基准数据集上的表现显著超越了所有监督和无监督的方法。
PhiGnet的泛化稳健性也得到了验证,能够处理与训练集中蛋白质序列同一性不同的蛋白质。当设置不同的最大序列同一性阈值(30%、40%、50%、70% 和 95%)时,随着序列同一性的增加,PhiGnet表现出更优的预测性能。
进化信息在PhiGnet中扮演着重要角色,不仅用于预测蛋白质功能注释,还能识别功能相关的位点。为此,我们进行了消融实验来评估EVC/RC对PhiGnet的贡献。实验结果表明,PhiGnet能够准确地为蛋白质分配功能注释。此外,利用EVC或RC的PhiGnet展示了学习序列功能关系的强大能力,其性能通常优于或至少等同于其他方法。
我们进一步探讨了PhiGnet从残基群落中识别出的功能相关残基中提取有意义特征的能力。通过计算残基的激活分数来突出它们对蛋白质功能的重要性。值得注意的是,预测的残基与实验测定确定的功能位点残基相吻合,并且比仅使用RC识别的效果更好。
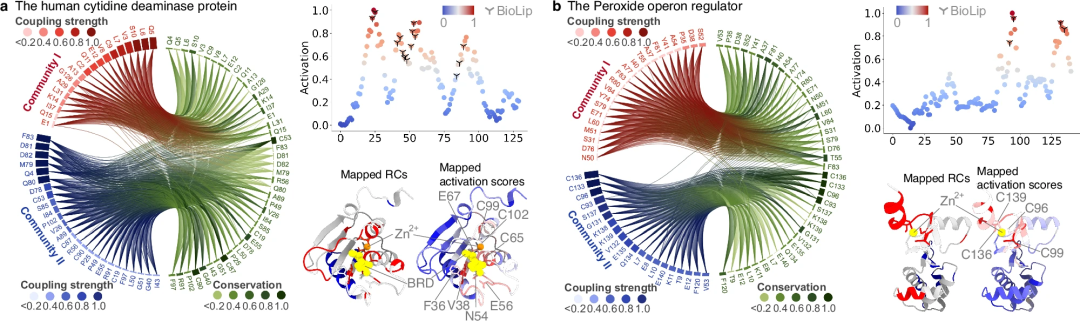
研究表明,进化信息,尤其是包含在RC中的信息,足以指定蛋白质的功能并定量表征功能位点的残基。此外,相较于EVC中较低层次的信息,RC包含了更高层次的进化知识。同时,RC中的信息对于提升PhiGnet在残基层面识别功能相关位点的能力至关重要。
总而言之,PhiGnet 的优越性能可归因于其有效利用了蛋白质序列的进化信息及其数据中的高阶模式,从而能够更深入且精确地理解蛋白质的功能。PhiGnet 的关键成功因素在于采用统计信息图卷积神经网络,以促进从大量序列数据集中提取的进化信息的层次化学习。这种方法在很大程度上超越了当前的监督与非监督方法,并有望指导未来的生物学及临床研究。
然而,PhiGnet 方法也存在局限性,特别是在序列多样性较低的蛋白质家族中可能出现偏差或噪声。将(共)进化信息整合进 PhiGnet 可能会干扰对残基群落的准确识别,尤其是当这些信息来源于高度保守的蛋白质家族时。尽管将物理提取的知识融入 PhiGnet 已经相较于其他方法实现了显著改进,但在解析 PhiGnet 内部的学习机制方面仍面临重大挑战。
进化数据与机器学习技术的协同作用将为准确预测和设计蛋白质的生物物理特性开辟新的途径。