

AI首度破解量子物理难题,DeepMind精准计算量子激发态,成果登Science
编辑日期:2024年08月24日

编辑 | KX
此前,谷歌旗下的DeepMind团队研发了一种名为费米子神经网络(FermiNet)的技术,特别擅长模拟大量电子的量子基态。
最初,FermiNet主要针对分子的基态进行研究。但在高能条件下,比如光照或高温,分子中的电子可能跃迁到更高能级——即所谓的激发态,这对于物理与化学领域都至关重要。
尽管如此,基于第一性原理来实现对激发态特性的准确、可扩展且稳定的计算仍然是一个重大的理论挑战。
如今,DeepMind的研究人员提出了一种新的计算激发态的方法,相比以往的技术更为强大且通用。这种方法不仅适用于FermiNet,也适用于其他的数学模型和神经网络。
新方法能够精确计算多种原子和分子的激发态,其准确性远超现有的深度学习计算方法(特别是在大型系统中),并且能够广泛应用于各类量子系统。
论文的第一作者及通讯作者David Pfau兴奋地表示:“这是深度学习首次成功而精准地解决了量子物理学中的一些最复杂问题。我们期待这将是迈向通用量子模拟深度学习方法的重要一步。”
相关研究成果以“利用神经网络准确计算量子激发态”为题,在《科学》杂志上发表!
论文链接:https://www.science.org/doi/abs/10.1126/science.adn0137 分子激发态
当分子及材料受到大量的能量刺激(例如光照射或高温)时,其电子会被激发进入一个暂时性的新状态,即所谓的激发态。
分子在不同状态间转变时所吸收和释放的确切能量值构成了每种分子和材料的独特“指纹”。这些特征直接影响了从太阳能电池板和LED灯到半导体和光催化剂等多种技术的表现。同时,这些过程也在诸如光合作用和视觉等生物过程中扮演着至关重要的角色。
然而,这一独特“指纹”的模拟极为复杂,因为激发电子遵循量子力学规律,意味着电子在分子中的位置始终处于不确定性之中,只能通过概率来描述。
FermiNet 已经能够在一系列包含不同性质的原子和小分子中生成极为精确的、甚至在某些情况下达到了前沿水平的基态能量。
最初,FermiNet 的研究重点集中在分子的基态上。但当分子和材料受到大量能量刺激(如光或高温),电子可能会跃升至更高能级,即激发态。
相较于基态能量的计算,激发态能量的精确计算更为艰难。即便是基态化学计算的金标准方法,如耦合簇方法,在处理激发态时也会出现数倍的误差。尽管研究者希望将 FermiNet 的应用扩展至激发态领域,现有的方法却未能达到与前沿技术相媲美的效果。
提出更强大、更通用的激发态计算新方法
DeepMind 开发了一种新的算法,该算法采用变分蒙特卡罗方法来估算量子系统的激发态,并且此算法不包含任何自由参数,也无需进行状态正交化,而是将问题转化为寻找扩展系统基态的问题。这种方法可以计算任意可观测量,包括非对角期望值,例如跃迁偶极矩。
该方法非常适合用于神经网络分析,通过结合 FermiNet 和 Psiformer ansatz,可以准确地计算一系列分子的激发能量和振荡器强度。
图示展示了从锂到氖的一行首个元素原子的激发态能量。这是应用 NES-VMC 方法于 FermiNet 所获得的结果。(来源:论文)
研究人员结合利用了神经网络 ansätze 的灵活性与数学洞见,他们将寻找系统激发态的问题转变为寻找扩展系统基态的问题,这一问题随后可以通过标准 VMC 方法解决。这种方法被称为自然激发态 VMC (NES-VMC)。
激发态之间的线性独立性是通过 ansatz 的函数形式自动保证的。每个激发态的能量以及其它可观测量都是通过对单态 ansätze 上的哈密顿期望值矩阵进行对角化得到的,这一过程可以累积计算并无需额外成本。
重要的是,这种方法不包含任何可调的自由参数,也无需使用惩罚项来确保正交性。研究者们采用两种不同的神经网络结构(FermiNet 和 Psiformer)来验证该方法的准确性。
从单个原子到苯
研究团队在其方法上进行了测试,范围涵盖了从单个原子到苯分子大小的基准系统。NES-VMC 在第一行原子上的表现证明了其准确性,其结果与实验数据高度吻合;对于一系列小分子,该方法同样获得了与当前最佳理论估计相当的高度精确的能量和振荡器强度。
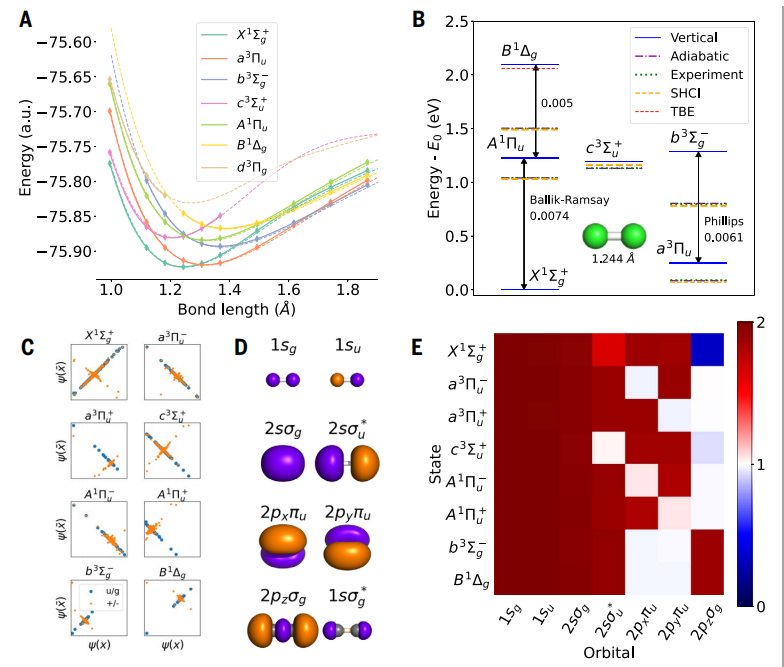
图示展示的是碳二聚体分子的激发态。(来源:论文)
在一种名为碳二聚体的小而复杂的分子上,NES-VMC 达到了 4 meV 的平均绝对误差 (MAE),这比之前的黄金标准计算准确度提高了五倍之多。
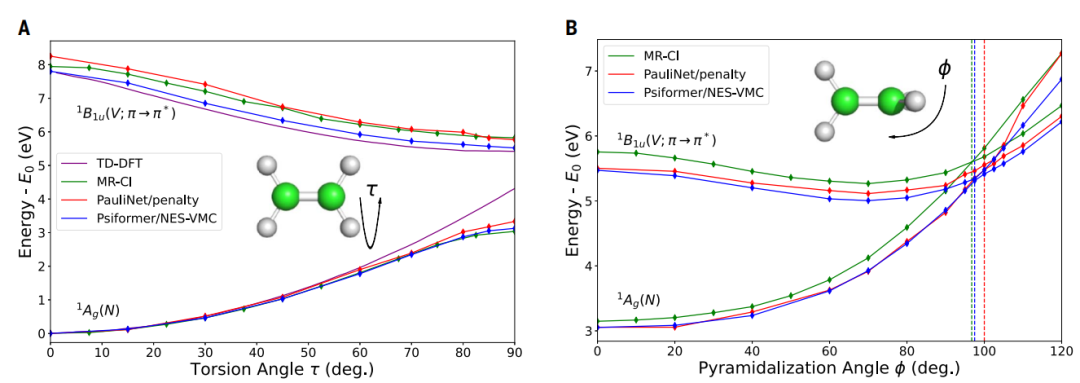
图示展示的是乙烯分子的激发态和圆锥交叉点。(来源:论文)
在处理乙烯分子时,NES-VMC 准确地描绘了扭曲分子的圆锥交叉,并且其结果与高精度的多参考组态相互作用 (MR-CI) 的计算结果高度吻合。
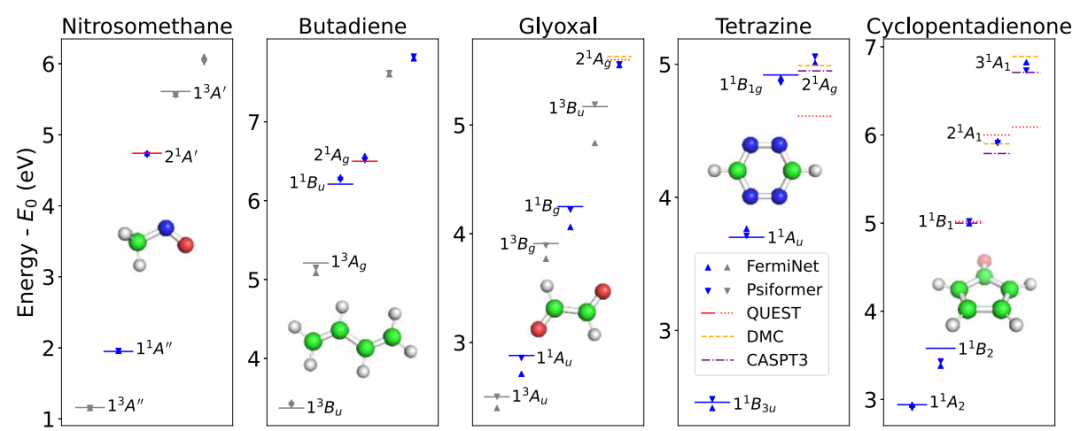
图示:较大双激发系统的激发态。(来源:论文)
研究还探讨了五个具有低位双激发的挑战性系统,其中包括多种苯类分子。在所有方法都能良好一致地描述垂直激发能的系统中,Psiformer 在各种状态下均达到了化学级别的准确性,包括丁二烯,在该分子中,某些状态的排序问题已经争论了几十年。
对于四嗪和环戊二烯酮这两个先前最先进的计算被证实不准确的案例,NES-VMC 的结果与近期复杂的扩散蒙特卡罗 (DMC) 及完全活性空间三阶微扰理论 (CASPT3) 的计算结果十分接近。
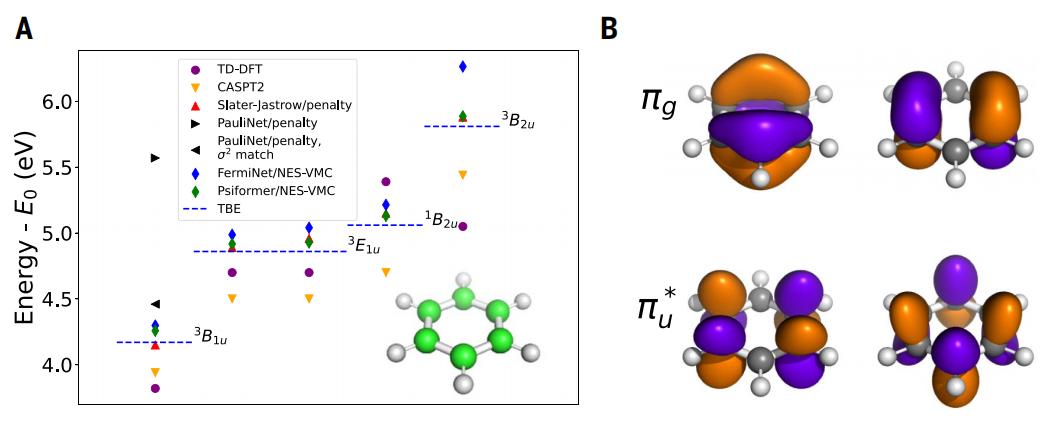
图示:苯的激发态。(来源:论文)
最后,研究还关注了苯分子。其中,NES-VMC 与 Psiformer ansatz 结合使用,相较于其他方法(包括带有惩罚项的神经网络 ansätze),它与理论最佳估计值的一致性更好。这一发现不仅验证了提出方法的数学正确性,而且表明神经网络能在现有计算方法的极限条件下准确地表征分子的激发态。
未来在多体量子力学中的应用前景
NES-VMC 是一种无需参数、数学上合理的激发态变分原理。将其与神经网络 ansätze 结合使用,能够在广泛的基准问题中达到显著的准确性。
对量子系统激发态进行精确的 VMC 计算为众多领域开辟了可能性,并极大地拓宽了神经网络波函数的应用范围。
尽管这项研究仅聚焦于分子体系中的电子激发态以及神经网络波函数试探函数(ansätze),NES-VMC 方法却能够适用于任意量子哈密顿量及任意试探函数,从而实现精准的计算研究。这种方法有助于加深科学家们对于振动电子耦合、光学带隙、核物理等复杂问题的理解。研究人员对此表示:“我们十分期待 NES-VMC 与深度神经网络在未来如何解决多体量子力学中最棘手的问题。”
参考资料:
https://x.com/pfau/status/1826681648597135464
https://deepmind.google/discover/blog/ferminet-quantum-physics-and-chemistry-from-first-principles/
https://www.imperial.ac.uk/news/255673/ai-tackles-most-difficult-challenges-quantum/